Neil Davies discusses a new paper on a genome-wide association study of almost 180,000 siblings and discusses what additional insight siblings bring to such studies.
Thousands of genome-wide association studies (GWAS) have been published, however, the vast majority have used samples of unrelated individuals. We have recently published a sibling GWAS published in Nature Genetics. In our study, we used almost 180,000 siblings across 19 studies from around the world. But why are siblings interesting for GWAS?
GWAS have already identified tens of thousands of single nucleotide polymorphisms (SNPs) related to phenotypes – using samples of unrelated individuals. However, correlation is not equal to causation. Increasing evidence suggests these associations can be driven by more than individual-level biological effects.
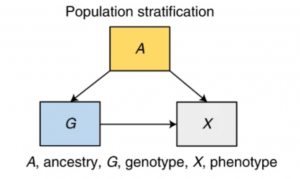
There can be three key sources of bias. The first potential bias is population stratification. This means the differences in the frequency of the genetic variants that relate to phenotypic differences. For example, Iron Brew consumption will associate with variants more common in Scotland. These associations are biased evidence of the causal effect of the variant on the phenotype!
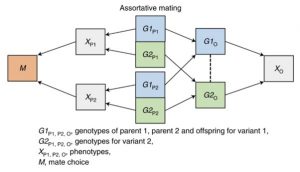
The second bias is assortative mating. People don’t mate at random. For example, studies have shown that more educated people tend to have more educated and taller partners. Such trends can result in biased associations between SNPs and phenotypes in the offspring.
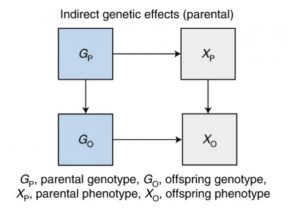
The third bias is indirect parental genetic effects (also known as dynastic effects).
In these, the genotype is expressed in parents, which in turn affects offspring outcomes. One example of this is that the education of parents may influence educational outcomes in the offspring, again biasing SNP-phenotype associations.
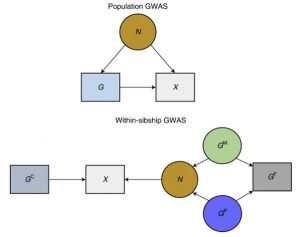
How can data from siblings help overcome these biases? Siblings inherit their genetic variants from their parents at random. They are nature’s randomized control trials. If the siblings who share the genotype have more similar trait measures, researchers can be more confident that the genotype is influencing the trait directly.
Looking at the differences between siblings controls for each of the sources of bias above.
Which phenotypes suffer most from these biases? In our Nature Genetics paper, we estimated the shrinkage from the population to sibling estimates for 25 phenotypes, to see which suffered most from these biases. We estimated this by looking at how much the associations shrunk between the population estimates (without comparing within siblings), to the within sibling estimates. The larger shrinkage in the LD-score regression plot below indicates more bias.
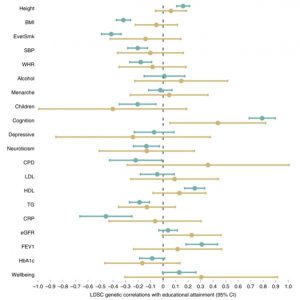
We found that previously reported genome-wide association study (GWAS) associations, which typically use more widely available population samples of unrelated individuals, tend to overestimate direct effects for many traits including educational attainment, cognitive ability, age when first gave birth, whether someone has ever smoked, depressive symptoms and number of children. We also found that estimates of heritability, genetic correlations and other genetic analysis methods could substantially differ when calculated using estimates from siblings.
Biases do affect genetic correlations
A major finding from our research was that these biases do affect genetic correlations. When we use sibling cohorts, the genetic correlations from LD-score regression between educational attainment and traits such as height and BMI are not detected. Note the change in power and precision in the plot below. This suggests that the correlations that are detected in population samples are unlikely to be due to a causal effect of the genetic variants in the individuals.
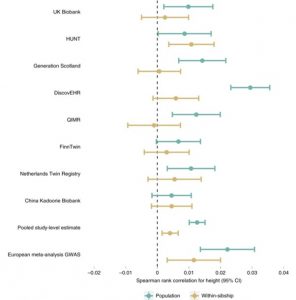
Are recent findings on polygenic adaption robust to these biases? Yes, height is likely to be under polygenic selection. This suggests that selective pressures in the human population have affected the number of height-associated alleles in the population. This could lead to changes in the average height of the population over multiple generations.
Are sibling samples “better” than “population” samples?
Whether sibling samples such as we use in our study are “better” than population studies depends on the question you want to look at. Large population-based samples of unrelated individuals are great if you want to discover new genetic variants associated with a disease or other outcomes, or are interested purely in prediction.
However, if you are interested in understanding why genetic variants associated with an outcome like height, BMI, or education, then family studies can provide a powerful source of evidence. In this paper, we only looked at a very small number of phenotypes, but these results suggest that these biases are more likely for social/behavioural phenotypes, and more biological ones are less likely to be biased.
What’s next? The international collaboration established for this study is continuing to work together and explore these issues further. The next steps include using bigger samples of siblings and estimating the relative contribution of these sources of bias using samples of parent-offspring trios.
A massive thanks to all our co-authors – an international group of 100 scientists were involved in this study – and many, many others. Amazing being able to work with you all!