
In July 2023 a group of researchers from IEU travelled to Entebbe, Uganda to deliver a short course on genome-wide association analyses and Mendelian randomization. Amanda Chong shares a glimpse of what happened during the week.
Blog posts on causality in population health from the MRC Integrative Epidemiology Unit
In July 2023 a group of researchers from IEU travelled to Entebbe, Uganda to deliver a short course on genome-wide association analyses and Mendelian randomization. Amanda Chong shares a glimpse of what happened during the week.
By Emma Anderson, Apostolos Gkatzionis, Lucy Goudswaard, Ruth Mitchell, Chin Yang Shapland, Kaitlin Wade and Venexia Walker
In recent years, there has been an explosion of research in Mendelian randomization (MR). It is a useful tool for inferring causality between exposures and outcomes of interest using genetic data. The Mendelian randomization conferences organised by the MRC Integrative Epidemiology Unit (IEU) regularly provide an opportunity to explore these issues.
In 2018, one of the conference attendees was Dr. Sarah Atkinson from Kemri Wellcome Trust in Kilifi, Kenya. As the conference progressed, she began chatting with Dr Ruth Mitchell and the idea emerged of IEU hosting an MR course for African scientists. Sarah applied and was awarded funding from the Wellcome Trust, which was supplemented by the IEU, to host six experts in MR from the IEU to teach a five-day course on Mendelian randomization to African researchers in Kilifi. (more…)
In two-sample Mendelian randomization (MR), a type of epidemiological method, we combine the results from different genetic studies to study the causal relationship between human characteristics and disease. For example, we might take results from a genetic study of smoking and a different genetic study of cancer. We can combine their results to understand whether smoking might be a cause of cancer. If the same position in the genome is associated with smoking in the first study and with cancer in the other study, this can provide evidence that smoking is a causal factor in cancer. However, it’s also possible that this position in the genome could be related to smoking and cancer via separate pathways. This phenomenon is known as “horizontal pleiotropy” and is a common source of bias in Mendelian randomization research.
Another, often under-appreciated, source of bias are errors in metadata. To understand this we need to understand what genetic results look like in practice. Below is an example of a genetic results file with 5 rows and 6 columns (a typical file might actually have several million rows).

Each row refers to a single position in the human genome that varies between people. These positions are referred to as “genetic variants” (also known as polymorphisms). The particular type of variant that an individual carries is known as their allele.
Below is an example of metadata. The metadata helps us understand the contents of the results file. It tells us what the columns represent.

Some columns in the results file will describe the relationship (“effect” column) between the genetic variant and some human characteristic (e.g. smoking) and there will be additional columns that help researchers interpret this relationship. These additional columns include things like the identity of the allele that is used to model the relationship (e.g. if people have allele “A” they may be more likely to smoke compared to people without this allele) or information on how common the allele is in the population. These columns are also known as the “effect allele” and “effect allele frequency” columns. Metadata errors refer to mistakes in how these columns are reported. For example, maybe allele1 is reported as the effect allele column when in fact it should have been allele 2 that is described in this way. Sometimes the information provided in metadata is ambiguous. For example, the metadata tells us that the “freq” column represents allele frequency but there are two alleles. Is this the frequency of allele1 or allele2? We can’t be sure. Another type of error refers to mistakes in the reported results, for example reporting that a genetic variant increases the probability that a person smokes when in reality it has no effect (in other words the effect is zero). This is known as a summary data error. Failure to identify these errors can lead to mistakes in Mendelian randomization analyses, such as finding that smoking protects against cancer (when we know the opposite is true).
These types of errors were fairly easy to avoid during the early years of Mendelian randomization research, when studies tended to be hypothesis-driven and focused on small numbers of relationships (although errors still occurred). Mendelian randomization study designs are, however, increasingly complex and hypothesis-free, sometimes assessing relationships amongst 100s or even 1000s of characteristics and diseases. New online platforms and databases that collate genetic results from many different sources, and provide tools that can automate analyses, make these studies easier to undertake than ever before. The downside is that they probably make meta and summary data errors more likely.
We address this issue in a new pre-print: “Design and quality control of large scale Mendelian randomization studies”. We present an R package and set of quality control tools that identify meta and summary data errors, which we developed for the Fatty Acids in Cancer Mendelian Randomization Collaboration (FAMRC). The FAMRC is a pan-cancer MR study that seeks to evaluate the causal relevance of fatty acids for risk of major cancers. We wanted to maximise the quality of the genetic study results we collected from the cancer studies, to ensure the integrity of our Mendelian randomization analyses. After implementing our tools, we found major meta and summary data errors in 7 (13%) of 55 genetic studies in the FAMRC.
The basic principle of our quality control approach is to identify errors through
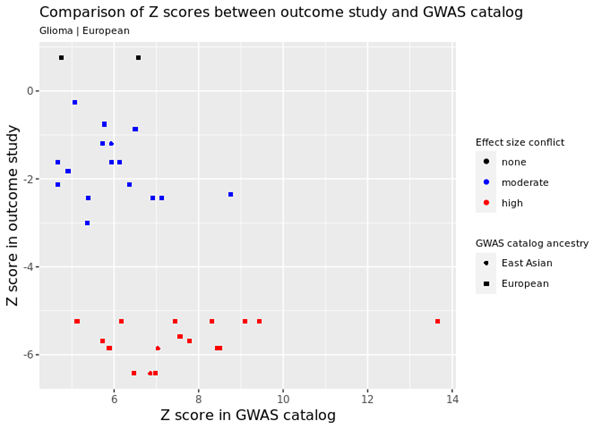
For example, we identified genetic variants that are known to cause cancer and checked that the same variants had the expected relationship in the FAMRC. In the figure below, every data point represents a single genetic variant that is known to increase cancer risk. The horizontal or X axis shows the known relationship in the GWAS catalog (this is a database of known genetic associations with 1000s of human characteristics in 1000s of genetic studies) and the vertical or Y axis shows the relationship in one of the studies in the FAMRC. Each axis shows the Z score, which is basically a standardised measure of how each genetic variant affects cancer risk (positive values mean that the variant increases risk of cancer and negative values indicate they decrease risk). As you can see, in the FAMRC study on the vertical Y axis, almost all the variants have negative Z values (indicating they reduce cancer risk), when in fact they are known to increase risk (the true relationship is represented by Z scores in the GWAS catalog). This discrepancy was caused by a metadata error, where the effect allele column was incorrectly labelled. We also found that the “frequency of the effect allele” was wrong. How common the allele is in the population was opposite to what we’d expect, based on comparison with other studies, confirming the presence of metadata errors.
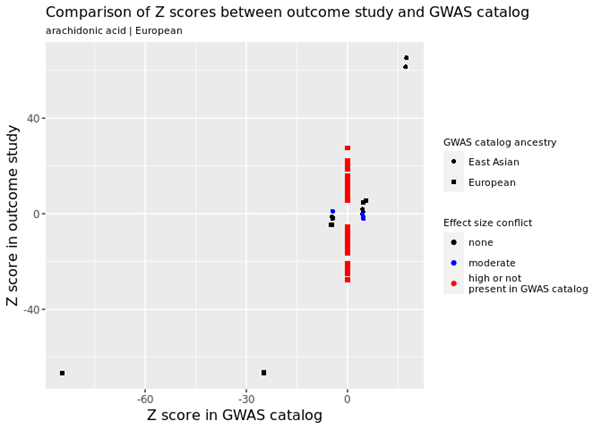
Various other types of errors were identified, including one study reporting that 100s of genetic variants had very strong effects on fatty acid levels when in fact they had no effect at all. For example, in the figure below, the many red data points refer to genetic variants in the FAMRC that had a very large effect on fatty acids but were not reported in the GWAS catalog, suggesting a potential problem with the genetic results.
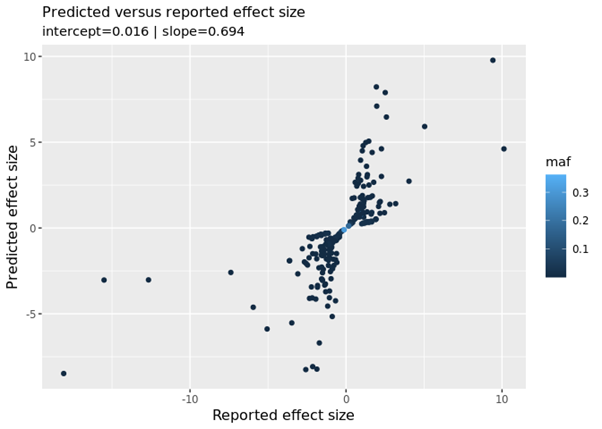
We also compared the reported results (how the genetic variants affected fatty acids in the FAMRC) to predicted results (how we would expect the genetic variants to affect fatty acids). In the figure below we see a “fanning-out” pattern, when what we should see is a strong linear relationship (i.e. the data points lying on a single straight line). This relationship can be summarised with the “slope” metric. We should see a slope of 1 (this means if the reported result increases by 1 the predicted result will also increase by 1), which is not the case. We confirmed with the data provider that low quality genetic variants had not been excluded from their study. Once the low quality variants had been excluded, the discrepancies disappeared.
When conducting Mendelian randomization analyses using results from genetic studies, researchers can avoid metadata and other errors by:
One issue we only partly addressed was the “two-sample assumption”: that the studies being compared come from the same population. In our own analyses, we found that the frequency of genetic variants was very similar across European-origin studies, indicating satisfaction of the assumption. On the other hand, our tools were not really optimised for this purpose. The need to assess the “same population” assumption is becoming more urgent with the growing diversity of genetic studies.
In conclusion, meta and summary data errors are an under-appreciated source of bias in MR research, especially in complex study designs. We developed an R package and set of tools that can be used to flag meta and summary data errors in the results of genetic studies, which in turn can be used to enhance the integrity of Mendelian randomization analyses. Our tools and methods are available to other researchers via the CheckSumStats R package.
Design and quality control of large-scale two-sample Mendelian randomisation studies
https://www.medrxiv.org/content/10.1101/2021.07.30.21260578v1
CheckSumStats package
https://github.com/MRCIEU/CheckSumStats
Amanda Hughes, Kaitlin H. Wade, Matt Dickson, Frances Rice, Alisha Davies, Neil M. Davies & Laura D. Howe
Follow Amanda, Kaitlin, Matt, Alisha, Neil and Laura on twitter
Young people with health problems tend to do less well in school than other students, but it has never been clear why. One explanation is that health problems directly damage educational outcomes. In that case, policymakers aiming to raise educational standards might want to focus first on health as a means of improving attainment.
But are there other explanations? What if falling behind in school can affect health, for instance causing depression? Also, many health problems are more common among children from less advantaged backgrounds – for example, from families with fewer financial resources, or whose parents are themselves unwell. These children also tend to do less well in school, for reasons that may have nothing to do with their own health. How do we know if their health, or their circumstances, are affecting attainment?
It is also unclear if health matters equally for education at all points in development, or particularly in certain school years. Establishing how much health does impact learning, when, and through which mechanisms, would better equip policymakers to improve educational outcomes.

Genetic data can help us answer these questions. Crucially, experiences like family financial difficulties, which might influence both a young person’s health and their learning, cannot change their genes. So, if young people genetically inclined to have asthma are more absent from school, or do less well in their GCSEs, that would strongly suggest an impact of asthma itself. Similarly, while falling behind in school might well trigger depression, it cannot change a person’s genetic propensity for depression. So, a connection between genetic propensity for depression and worse educational outcomes supports an impact of depression itself. This approach, of harnessing genetic information to better understand causal processes, is known as Mendelian randomization.
To find out more, we investigated links between
We used data from 6113 children born in the Bristol area in 1991-1992. All were participants of the Avon Longitudinal Study of Parents and Children (ALSPAC), also known as Children of the 90s. We focused on six different aspects of health: asthma, migraines, body mass index (BMI), and symptoms of depression, of attention-deficit hyperactivity disorder (ADHD), and of autism spectrum disorder (ASD). These conditions, though diverse, have two important things in common: they affect substantial numbers of young people, and they are at least in part influenced by genetics.
Alongside questionnaire data and education records, we also analysed genetic information from participants’ blood samples. From this information, we were able to calculate for each young person a summary score of genetic propensity for experiencing migraines, ADHD, depression, ASD, and for having a higher BMI.
We used these scores to predict the health conditions, rather than relying just on reports from questionnaire data. In this way, we avoided bias due to the impact of the young people’s circumstances, or of their education on their health rather than vice versa.
We found that, for each extra day per year of school missed in year 10 or 11, a child’s total GCSE points from their best 8 subjects was a bit less than half (0.43) of a grade lower. Higher BMI was related to increased school absence & lower GCSE grades.
Using the genetic approach, we found that young people genetically predisposed towards a higher BMI were more often absent from school, and they did less well in their GCSEs. A standard-deviation increase* in BMI corresponded to 9% more school absence, and GCSEs around 1/3 grade lower in every subject. Together, these results indicate that increased school absence may be one mechanism by which being heavier could negatively impact learning. However, in other analyses, we found a substantial part of the BMI-GCSEs link was not explained by school absence. It’s unclear which other mechanisms are at play here, but work by other researchers has suggested that weight-related bullying, and negative effects of being heavier on young people’s self-esteem, could interfere with learning.
*equivalent to the difference between the median (50th percentile) in population and the 84th percentile of the population
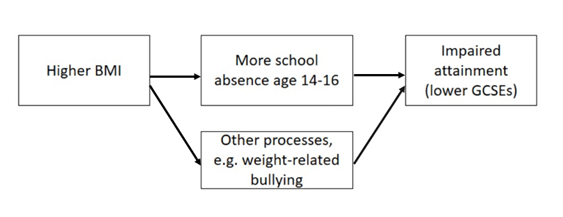
In line with previous research, young people genetically predisposed to ADHD did less well in their GCSEs. Interestingly, they did not have increased school absence, suggesting that ADHD’s impact on learning works mostly through other pathways. This is consistent with previous research highlighting the importance of other factors on the academic attainment of children with ADHD, including expectations of the school environment, teacher views and attitudes, and bullying by peers.
Our genetic analyses found little support for a negative impact of asthma, migraines, depression or ASD on educational attainment. However, we know relatively little about the genetic influences on depression and ASD, especially compared to the genetics of BMI, which we understand much better. This makes genetic associations with depression or ASD difficult to detect. So, our results should not be taken as conclusive evidence that these conditions do not affect learning.
Our findings provide evidence of a detrimental impact of high BMI and of ADHD symptoms on GCSE attainment, which for BMI was partially mediated by school absence. When students sent home during the pandemic eventually return to school, the impact on their learning will have been enormous. And while all students will have been affected, our results highlight that young people who are heavier, who have ADHD, or are experiencing other health problems, will likely need extra support.
Hughes, A., Wade, K.H., Dickson, M. et al. Common health conditions in childhood and adolescence, school absence, and educational attainment: Mendelian randomization study. npj Sci. Learn. 6, 1 (2021). https://doi.org/10.1038/s41539-020-00080-6
A version of this blog was posted on the journal’s blog site on 21 Jan 2021.
Amanda Hughes, Senior Research Associate in Epidemiology: amanda.hughes@bristol.ac.uk


 Yoonsu Cho, Bryony Hayes and Daniel Lawson
Yoonsu Cho, Bryony Hayes and Daniel Lawson
As we usher in the era of precision medicine – healthcare tailored to the individual – genetic information is being used to design drugs, tests and medical procedures. While this approach enables physicians to better predict the needs of patients and quickly adopt the most suitable treatment, it should be acknowledged that what is suitable for many, is not suitable for all. Appropriate medical care is linked with ancestry – for example, healthy people with African ancestry naturally exhale less air than reference samples of Europeans, leading to mis-diagnosis for Asthma. For people from Black and Minority Ethnicities, the potential impact of ethnicity is intensely debated in Cancer treatment. Research suggests that lack of BME participation in medical research will lead to poor medication choices, genetic tests being less useful, and any COVID-19 treatments being less well tested.
The World Health Organization has stated that “everyone should have a fair opportunity to attain their full health potential and that no one should be disadvantaged from achieving their potential”. However, systematic discrimination & socio-economic-related disadvantages such as lower education and difficulty accessing high quality jobs are overwhelmingly experienced by non-white people, with statistics showing that 80% of Black African and Caribbean communities are living in England’s most deprived areas (as defined by the Neighbourhood Renewal Fund). These factors contribute to people from those communities receiving worse medical care overall. Beyond this, poor representation in research now can only lead to systematically poorer healthcare in the years to come. In 2009, only 4% of genetic association studies used samples with non-European ancestry. Whilst this rose to almost 20% by 2016, this improvement was largely due to East Asian nations such as Korea, China and Japan initiating their own biobank projects, leaving many ethnicities under-represented. Hence, from a medical genetics perspective, “Black and Minority Ethnicity” (BME) is well defined as “ethnicities without a rich nation to back a representative genetic biobank” and includes African ancestry.

Epidemiological comparisons – that is, comparing large numbers of people who develop disease and those that do not – often rely on genetics to infer which behaviours and conditions are causes and which are effects. These analyses use a technique called Mendelian Randomization (MR). MR has demonstrated, for example, that alcohol consumption causally increases body mass and made it clear that even moderate alcohol intake has no beneficial effect on health outcomes. Causal hypotheses are a critical pathway to drug discovery and public health intervention, but are based almost entirely on European populations. Since there are many genes that affect most disease risk and these are of different importance across ancestries, we cannot be certain that the associations found apply to other populations. This urgently needs to be addressed in order to:
Some populations have increased risk from specific diseases, and many people have ancestry from all over the world, making the categorisation of ‘race’ in medicine of some value but increasingly problematic. The IEU leads work on measuring this ancestry variation, which is important for individuals’ health. Getting at the cause of disease is key for understanding the effects of genes on disease risk and traits. Data on varied ethnicities is valuable for science, simply by showing us more variation. Traits such as height, weight and pre-inclination for education may not be directly related to ethnicity, but data from varied ancestries still helps to separate genetic cause from effect. Paradoxically, the least available data on African ancestry is particularly valuable scientifically, due to the lack of variation in the population that came out-of-africa around 50,000 years ago.
Acknowledgement of this deficit is becoming more widespread, and the Black Lives Matter movement has refocused attention on representation in science, but the solution remains undetermined. How do we in the science and research community push for better diversity and representation in our resources? Biobanks operate on a consensual ‘opt in, opt out’ system and tend to favour certain groups. In 2016 the Financial Times generalised the participants of UK Biobank and “healthy, wealthy and white”, but why do so many more individuals from this demographic ‘opt in’? In 2018 Prictor et al theorised that BME groups may experience more barriers to participation such as location, cultural sensitivities around human tissue, and issues of literacy and language. However, given the history of the relationship between the research community and minority groups, seen in cases such as the Tuskegee Study, it is easy to see why BME populations might be less inclined to participate, if invited.
Although there is still need for considerable change, several recent developments will help, including the China Kadoori Biobank, the ancestrally diverse US-based Million Veterans program, and many others. However, given restrictions on privacy and reporting methods, these biobanks are hard to compare. Currently the IEU is part of a multi-national effort to develop tools to get the best science possible out of these comparisons, whilst simultaneously respecting privacy and data security issues. The IEU has been collaborating with various research groups across the world to make our research more reproducible. Building tools that work at scale is a challenge encompassing Mathematics, Statistics, Computer Science, Engineering, Genomics and Epidemiology, but this work is paving the way to promoting representative research that is inclusive and applicable for all.




 Deborah Lawlor, Professor of Epidemiology, Emma Anderson, MRC Research Fellow, Marcus Munafò, Professor of Experimental Psychology, Mark Gibson, PhD student, Rebecca Richmond, Vice Chancellor’s Research Fellow
Deborah Lawlor, Professor of Epidemiology, Emma Anderson, MRC Research Fellow, Marcus Munafò, Professor of Experimental Psychology, Mark Gibson, PhD student, Rebecca Richmond, Vice Chancellor’s Research Fellow
Follow Deborah, Marcus, and Rebecca on Twitter
Sleep is important for health. Observational studies show that people who report having sleep problems are more likely to be overweight, and have more health problems including heart disease, some cancers and mental health problems.
A major problem with conventional observational studies is that we cannot tell whether these associations are causal; does being overweight cause sleep problems, or do sleep problems cause people to become overweight? Alternatively, factors that influence how we sleep may also influence our health. For example, smoking might cause sleep problems as well as heart disease and so we are fooled (confounded) into thinking sleep problems cause heart disease when it is really all explained by smoking. In the green paper Advancing our Health: Prevention in the 2020s, the UK Government acknowledged that sleep has had little attention in policy, and that causality between sleep and health is likely to run in both directions.
But, how can we determine the direction of causality for sure? And, how do we make sure we are results are not confounded?
Our genes are randomly allocated to us from our parents when we are conceived. They do not change across our lifespan, and cannot be changed by smoking, overweight or ill health.
Here at the MRC Integrative Epidemiology Unit we have developed a research method called Mendelian randomization, which uses this family-level random allocation of genes to explore causal effects. To find out more about Mendelian randomization take a look at this primer from the Director of the Unit (Prof George Davey Smith).
In the last two years, we and colleagues from the Universities of Manchester, Exeter and Harvard have identified large numbers of genetic variants that relate to different sleep characteristics. These include:
We can use these genetic variants in Mendelian randomization studies to get a better understanding of whether sleep characteristics affect health and disease.
In our initial studies we used Mendelian randomization to explore the effects of sleep duration, insomnia and chronotype on body mass index, coronary heart disease, mental health problems, Alzheimer’s disease, and breast cancer. We analysed whether the genetic traits that are related to sleep characteristics – rather than the sleep characteristics themselves – are associated with the health outcomes. We combined those results with the effect of the genetic variants on sleep traits which allows us to estimate a causal effect. Using genetic variants rather than participants’ reports of their sleep characteristics makes us much more certain that the effects we identify are not due to confounding or reverse causation.

Our results show a mixed picture; different sleep characteristics have varying effects on a range of health outcomes.
Having better research evidence about the effects of sleep traits on different health outcomes means that we can give better advice to people at risk of specific health problems. For example, developing effective programmes to alleviate insomnia may prevent coronary heart disease and depression in those at risk. It can also help reduce worry about sleep and health, by demonstrating that some associations that have been found in previous studies are not likely to reflect causality.
If you are worried about your own sleep, the NHS has some useful guidance and signposting to further support.
Deborah A Lawlor mailto:d.a.lawlor@bristol.ac.uk
This research has been published in the following open access research papers:
Genome-wide association analyses of chronotype in 697,828 individuals provides insights into circadian rhythms. Nature Comms (2019) https://www.nature.com/articles/s41467-018-08259-7
Biological and clinical insights from genetics of insomnia symptoms. Nature Gen. (2019) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6415688/
Genome-wide association study identifies genetic loci for self-reported habitual sleep duration supported by accelerometer-derived estimates. Nature Comms. (2019) https://www.nature.com/articles/s41467-019-08917-4
Investigating causal relations between sleep traits and risk of breast cancer in women: mendelian randomisation study. BMJ (2019) https://www.bmj.com/content/365/bmj.l2327
Is disrupted sleep a risk factor for Alzheimer’s disease? Evidence from a two-sample Mendelian randomization analysis. https://www.biorxiv.org/content/10.1101/609834v1 (open access pre-print)
Evidence for Genetic Correlations and Bidirectional, Causal Effects Between Smoking and Sleep Behaviors. Nicotine and Tobacco (2018) https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6528151/
 Dr Philip Haycock
Dr Philip Haycock